Автор: Дмитрий Юрьевич Блохин,
доктор медицинских наук,
зав. лабораторией фармакоцитокинетики НИИ экспериментальной диагностики и терапии опухолей ГУ РОНЦ им. Н. Н. Блохина РАМН
Эта статья — о причинах возникновения, закономерностях развития и путях лечения онкологических болезней, а также о тех трудностях, с которыми сталкиваются ученые-онкологи при разработке новых средств и методов лечения рака. Но вначале стоит вспомнить некоторые основные понятия.
Несколько слов про опухолевые клетки
Человеческий организм состоит примерно из 100 триллионов клеток. Изменение этого количества всегда физиологически оправданно. Например, при воспалении увеличивается число белых клеток крови (лейкоцитов), которые противостоят возбудителям инфекции. При интенсивных физических нагрузках возрастают количество мышечных клеток и мышечная масса. Процесс поддержания оптимальной численности клеток — клеточный гомеостаз — осуществляет сложная система контроля клеточных делений (пролиферации) и клеточной гибели.
Каждая клетка имеет свою продолжительность жизни: эритроциты — около 120 дней, лейкоциты — от нескольких часов (нейтрофилы) до нескольких недель (лимфоциты), а «клетки памяти», как называют специализированные иммунные лимфоциты, могут жить десятки лет. По истечении отпущенного ей срока клетка погибает. Гибель эта упорядоченна и генетически запрограммированна. Программа клеточной гибели включается, если клетка больше не нужна организму (например, принадлежит эмбриональной ткани), состарилась, заразилась вирусом, накопила много мутаций или получила иное не подлежащее восстановлению повреждение. При этом происходит последовательная саморазборка клетки на фрагменты, которые затем поглощают макрофаги или соседние клетки в качестве питательного и строительного субстрата. Как правило, в литературе для обозначения программированной клеточной гибели используют термин «апоптоз».
Программа клеточной гибели срабатывает только после многократного подтверждения «сигнала смерти». Сигнал может прийти из окружающей клетку среды или от собственных внутриклеточных «датчиков неблагополучия». Внешний сигнал клетка воспринимает специальными «рецепторами смерти», находящимися на ее поверхности. Существуют и различные внутренние сигналы, возникающие при неустранимых внутренних повреждениях клетки (в большинстве случаев — молекул ДНК), которые препятствуют ее нормальному делению или функционированию. Но независимо от источника и места получения этого сигнала в итоге запускается один и тот же каскад активации «суицидных» ферментов, которые и завершают выполнение программы: эффекторные каспазы, ДНК-фрагментирующий фактор и др.
В здоровом и нормально функционирующем организме ежесекундно погибает огромное количество клеток, столько же образуется вновь. Но иногда процесс клеточного гомеостаза выходит из-под контроля и возникает опухоль.
Опухолью называют патологическое разрастание ткани, состоящее из качественно измененных (атипичных) по морфологии, степени дифференцировки и характеру роста клеток. Не всякое увеличение объема ткани представляет собой опухоль. Отек, например, связан не с разрастанием клеток, а с накоплением межклеточной жидкости, гипертрофированные мышцы культуриста — адаптация организма к длительным физическим нагрузкам. Эти изменения преходящи: после снижения мышечных нагрузок дополнительная ткань подвергается инволюции (то есть рассасывается). Появление опухоли с адаптацией не связано, и инволюции она не подвержена. Опухоль, в отличие от нормальной ткани, не имеет выраженной структуры, ее строение в той или иной степени беспорядочно. Она образована клетками, которые не завершают дифференцировку и несут признаки юных, а часто и эмбриональных форм.
Если разрастание опухоли ограничивается местом ее возникновения, то она доброкачественная. К доброкачественным опухолям относятся миомы, липомы, эпителиомы, аденомы (окончание -ома обозначает «опухоль», а корень слова часто происходит от названия ткани, из которой возникли опухолевые клетки), папилломы, полипы, пигментные невусы — «висячие родинки», бородавки и многие другие. Доброкачественные опухоли, как правило, не представляют угрозы жизни больного, поскольку носят локальный характер. Исключение составляют опухоли мозга, которые в силу жестко ограниченного пространства черепной коробки могут механически сдавливать соседние участки мозга и кровеносных сосудов, вызывая парезы, параличи и даже гибель больного.
Если рост опухоли не ограничен собственной тканью и органом, а оторвавшиеся от основного узла атипичные клетки мигрируют в соседние и отдаленные органы, вызывая появление там вторичных опухолевых узлов (метастазов), то такая опухоль злокачественна.
Помимо способности образовывать метастазы, то есть существовать вне привычного клеточного окружения, для раковых клеток характерно неуправляемое деление, причем делиться они могут неограниченное количество раз, не обнаруживая при этом признаков старения, и в значительной мере утрачивают способность к программированной клеточной гибели. Именно совокупность всех этих признаков и отличает раковую клетку от нормальной.
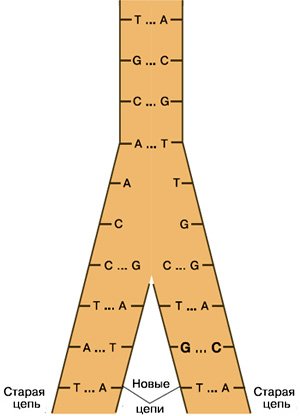
Рис. 1. Клетка, получившая мутацию, передает ее одной из дочерних клеток. Изображение: «Химия и жизнь»
Опухолевая трансформация клетки происходит, когда она накапливает некоторое количество мутаций, причем не любых, а критических для канцерогенеза. Пока ученые точно не знают, сколько мутаций и в каких именно генах должно произойти, чтобы клетка стала опухолевой. Очевидно, никак не меньше пяти, а по самым оптимистическим прогнозам 8–10. Важно, что речь идет не о каком-то определенном наборе мутаций: их комбинации, определяющие опухолевую трансформацию, могут быть самыми разными. С молекулярно-генетической точки зрения не существует двух совершенно одинаковых опухолей, как и совершенно одинаковых причин их возникновения. Уникальность каждой опухоли намного превышает уникальность дактилоскопических узоров.
«Универсальной» или «главной» мутации, необходимой и достаточной для превращения нормальной клетки в раковую, ученые не обнаружили. Однако об одном гене, изменения в котором часто приводят к злокачественной трансформации, стоит упомянуть. Называется этот ген ТР53, а его белковый продукт р53 (такое невыразительное обозначение произвели от «протеин с молекулярной массой 53 килодальтона») регулирует активность более 150 генов, контролирующих цикл клеточного деления.
Процесс клеточного деления очень сложен и таит в себе немало опасностей, связанных с возникновением и закреплением соматических мутаций, то есть мутаций, возникающих в соматических клетках. Чтобы избежать такой беды, в организме существует система генетического самоконтроля клеток. Известно по крайней мере четыре контрольные (или сверочные) точки, в которых происходит анализ правильной последовательности событий репликативного цикла. Если что-то прошло не так, то пролиферация временно останавливается, а если повреждение не удается исправить, включается программа клеточной гибели, которая не позволит мутантным клеткам размножаться. Ключевую роль в этом процессе играет белок р53, который часто именуют «стражем генома», а постоянно функционирующий ген ТР53 относят к опухолевым супрессорам (тормозящим развитие опухолей). Но насколько он важен для опухолевой супрессии, пока неясно. С одной стороны, возникновение инактивирующих мутаций в гене ТР53 или полное прекращение его экспрессии (нокаут гена) вызывают дестабилизацию генома: формируется так называемый мутаторный фенотип клетки, при котором частота появления и накопления мутаций резко возрастает. Если мутация гена ТР53 получена по наследству от родителей, она присутствует во всех клетках организма и сопровождается развитием синдрома Ли-Фраумени, при котором еще в детстве возникают множественные опухоли. Такие пациенты редко доживают до совершеннолетия. Однако, как показали масштабные генетические исследования, проведенные в лабораториях разных стран, лишь чуть более половины всех исследованных злокачественных опухолей человека различной локализации и стадии развития несут мутации в гене ТР53; клетки же второй половины исследованного массива синтезируют нормальный белок р53, что, впрочем, не мешает им быть злокачественными!
Ежедневно в человеческом организме возникают сотни тысяч мутантных клеток. Их постоянно отслеживают и уничтожают две системы контроля: система клеточного генетического самоконтроля, о которой шла речь выше, и система неспецифического противоопухолевого иммунитета.
Система противоопухолевого иммунитета распознает мутантные клетки по наличию на их поверхности постороннего, не свойственного данному организму антигена или по отсутствию одного из абсолютно необходимых. К первым относятся так называемые опухоле-ассоциированные и вирусные антигены, а ко вторым — антигены главного комплекса гистосовместимости I класса, несущие информацию: «Я — свой». Если эти антигены не представлены на клетке, ей немедленно делает «смертельную инъекцию» клетка-киллер, которая осуществляет иммунологический надзор. Она формирует в стенке клетки-мишени канал, через который впрыскивает ферменты-гранзимы. Гранзимы «включают» проферменты класса каспаз — это основные исполнители программы клеточной гибели.
Исполнительный механизм системы противоопухолевого иммунитета сопряжен с механизмом обеспечения генетического самоконтроля. Это означает, что клетка, которая в результате мутации станет невосприимчива к действию одной системы контроля, будет неуязвима и для другой. Потомки такой клетки унаследуют приобретенный признак и положат начало формированию мутантного клона — способность ускользать от системы генетического самоконтроля позволит и в дальнейшем избегать гибели при тиражировании вновь появившихся мутаций. Эти клетки еще нельзя назвать опухоле-трансформированными, поскольку они пока не приобрели всех необходимых для этого генетических дефектов, но начало положено: мутаторный фенотип открыл простор для дальнейшего накопления мутаций.
Поскольку процесс мутагенеза носит случайный характер, в каждой клетке мутантного клона возникнет индивидуальный набор мутаций, и происходит клональное расщепление популяции. Появление новых мутаций отразится на фенотипе потомков — они будут постепенно утрачивать родительские черты, но приобретать новые свойства, в том числе те, которые присущи опухолевым клеткам. Наиважнейшее из них — способность к неограниченному числу делений, или репродуктивное бессмертие. Без этой способности все прочие приобретенные «опухолевые» свойства не будут представлять опасности: совершив положенное число удвоений, клетки необратимо утратят способность к делению — рост опухоли остановится, за чем последует ее постепенное саморазрушение. Если же клетка достигнет репродуктивного бессмертия, приобретение прочих опухолевых черт — только вопрос времени.
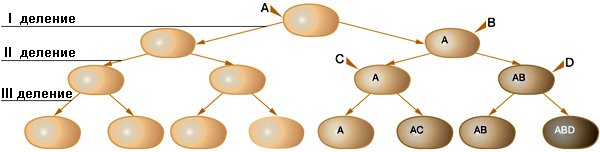
Рис. 2. Схема клонального расщепления потомства мутантной клетки. А, В, С и D — случайно возникшие мутации. Изображение: «Химия и жизнь»
Бывают случаи, когда возникшая доброкачественная опухоль в ходе своего роста по тем или иным причинам становится злокачественной — «малигнизируется». Так на месте доброкачественного пигментного невуса может образоваться меланома — одна из самых злокачественных опухолей кожи, как правило, образующая множественные метастазы. Малигнизация доброкачественной опухоли — процесс не обязательный, большинство таких новообразований существуют в организме годами, растут медленно и в основном доставляют лишь косметические неудобства. Однако злокачественная опухоль может развиться не только из доброкачественной, но и из совершенно здоровой ткани. В этих случаях появлению опухоли обычно предшествует «предрак» — компактное скопление измененных по морфологии мутантных клеток. Их потомки могут перерастать во внутритканевой, «местный» рак, который затем распространяется и образует инфильтрирующие злокачественные образования. Так происходит прогрессия опухолевого процесса, направление которой во всех случаях одинаково — от плохого к худшему.
Возникнув, опухолевая ткань не только безудержно растет вследствие бесконтрольного деления составляющих ее клеток, но и постоянно эволюционирует, порождая новые клеточные клоны, наиболее злокачественные из которых, то есть лучше приспособленные к автономному существованию, в процессе конкурентной борьбы вытесняют менее злокачественные. Остановить такую экспансию можно, лишь удалив опухоль из организма или, по крайней мере, ограничив ее рост.
Лечение онкологических заболеваний
Сегодня существует три основных метода лечения раковых больных: хирургическое удаление опухолевых узлов, химиотерапия и радиолучевая терапия, причем в подавляющем большинстве случаев их приходится комбинировать.
Хирургическое вмешательство эффективно лишь тогда, когда процесс локализован и иссечение опухоли в пределах здоровых тканей не разрушает функционирование жизненно важных органов. В иных случаях, а также если первичный опухолевый очаг вовсе отсутствует, например, при лейкозах, применяют химиотерапию, которая теоретически должна поражать опухолевые клетки вне зависимости от их локализации.
Идею «химиотерапии рака» впервые сформулировал Пауль Эрлих в начале XX века. Однако сложности проблемы избирательного поражения опухолевых клеток без вреда для клеток нормальных вынудили Эрлиха отказаться от практической реализации идеи. И только в конце 40—начале 50-х годов ушедшего столетия медики обнаружили химические соединения, которые не только останавливают деление и вызывают гибель опухолевых клеток в культуре, но и тормозят рост опухолей в организме. Первым официальным лекарством от рака стал эмбихин, впервые примененный на человеке в 1946 году. Созданный на основе иприта, боевого отравляющего вещества времен Первой мировой войны, эмбихин положил начало целому семейству противоопухолевых лекарств алкилирующего типа, применяющихся и поныне. За более чем полувековую историю своего существования химиотерапия выделилась в самостоятельную область клинической онкологии. Однако несмотря на значительные успехи в этой области, полного излечения с помощью одной химиотерапии удается добиться лишь при ограниченном круге опухолевых заболеваний, высокочувствительных к лекарственным препаратам: хорионэпителиоме матки, герминогенных опухолях яичка, лимфогранулематозе, лимфоме Беркита, остром лимфобластном лейкозе у детей. При химиотерапевтическом лечении больных саркомой Юинга, лимфосаркомами, аденокарциномами молочной железы и яичника, раком мочевого пузыря и некоторыми другими нозологическими формами химиотерапия позволяет получить значительный клинический эффект, но полностью излечиваются не более 10% больных. Еще скромнее выглядят результаты химиотерапии при лечении рака желудка, рака толстой кишки, немелкоклеточного рака легкого, а злокачественные опухоли пищевода, печени, поджелудочной и щитовидной желез, рак почки и рак шейки матки проявляют значительную устойчивость к лекарственному лечению. Тем не менее использование химиотерапевтических препаратов при комплексном лечении этих опухолей оправдано, поскольку позволяет после удаления опухоли подавить рецидивы заболевания и развитие метастазов, а в предоперационном периоде помогает уменьшить размер опухоли и облегчить ее хирургическое иссечение.
Причина столь невысокой клинической эффективности лекарственных методов лечения опухолей заключается в том, что полного исцеления можно достичь, только поразив все без исключения опухолевые клетки. Требования к лечению инфекционных и паразитарных заболеваний не столь суровы — уцелевших после лечения паразитов «зачищает» иммунная система. А раковые клетки организму «родные», иммунная система на них не реагирует, и если уцелеет хотя бы тысячная их часть, опухоль восстановится до начальных размеров всего через 10 делений (210 = 1024). Здесь и кроется основная трудность: истребить молниеносным ударом все опухолевые клетки не удается, а в процессе длительного лечения происходит не только восстановление популяции, но и ее прогрессия, с изменением свойств и спектра лекарственной чувствительности — всегда по направлению «от плохого к худшему». Так что химиотерапия наиболее эффективна на относительно ранних стадиях развития опухоли.
Открытие каждого нового класса химических соединений, обладающих противоопухолевой активностью, вызывало всплеск оптимизма, но всякий раз результаты оказывались значительно скромнее ожиданий. Первые лекарства от рака либо химически повреждали молекулы ДНК и белков (алкилирующие соединения: эмбихин, мелфалан, метилнитрозомочевина, циклофосфамид и др.), либо препятствовали процессу удвоения нити ДНК (антиметаболиты, первые из которых, метотрексат и 5-фторурацил, созданные в 1949 и 1956 годах соответственно, до сих пор применяют в онкологии). Позднее появились препараты, поражающие другие внутриклеточные мишени: противоопухолевые антибиотики (доксорубицин, блеомицин), вещества растительного происхождения (винбластин, паклитаксел, этопозид), комплексные соединения платины (цисплатин, карбоплатин). Несмотря на то что эти химические соединения действуют в клетках на самые разные молекулярные мишени, их объединяет способность избирательно подавлять рост и вызывать гибель опухолевых клеток при относительно малом повреждении клеток нормальных тканей. Параллельно с поиском новых противоопухолевых препаратов шло изучение молекулярных механизмов действия на клетку уже найденных и применявшихся на практике лекарств. По мере развития представлений о механизмах противоопухолевой активности разных препаратов стало очевидным, что вопрос о низкой эффективности химиотерапии опухолей неразрывно связан с другим, не менее актуальным. По словам академика Н. Н. Трапезникова, многие годы возглавлявшего Онкологический научный центр после Н. Н. Блохина, если раньше онкологи ставили вопрос, почему не действуют лекарственные препараты, то сейчас вопрос ставится иначе: а почему они действуют? Ответ на последний вопрос был найден совсем недавно.
Большинство противоопухолевых препаратов «первой волны» было отобрано в результате экспериментального поиска химических соединений, убивающих преимущественно опухолевые клетки (их называют веществами с потенциальной противоопухолевой активностью). Для этого ученые исследовали, как действуют на культуры раковых клеток миллионы природных и синтетических веществ. Этот метод называется методом случайного отбора, по-научному — рандомизированным скринингом. Далеко не каждое из отобранных соединений может впоследствии стать лекарством. Позднее ученые специально синтезировали химические соединения, которые теоретически должны ингибировать те или иные ферменты, важные для процесса клеточного деления. В результате этих двух подходов к поиску лекарств и был создан весь современный арсенал противоопухолевых средств.
Однако избирательность химиопрепаратов не абсолютна: в процессе лечения они наряду с опухолевыми часто поражают нормальные клетки, в первую очередь быстро обновляющихся тканей: костного мозга, эпителия желудочно-кишечного тракта и волосяных фолликулов кожи. Но если поражение фолликулов вызывает только облысение — досадный, однако временный косметический дефект, то массовая гибель клеток эпителия и костного мозга представляет реальную угрозу жизни пациентов.
Эффективность консервативных методов лечения рака до настоящего времени ограничена не только побочным токсическим действием на клетки нормальных тканей, но и лекарственной устойчивостью опухолей. Подавляющее большинство противоопухолевых природных и синтетических химических соединений действует на клетки непосредственно, проникая в них и поражая многообразные внутриклеточные молекулярные мишени. Ранее медики полагали, что противоопухолевые препараты вызывают в клетке несовместимые с жизнью химические повреждения биомакромолекул — в первую очередь нуклеиновых кислот и белков. Однако по мере развития наших представлений о механизмах программируемой клеточной гибели становилось очевидным, что практически все противоопухолевые лекарства от препаратов «первой волны» (эмбихин, 5-фторурацил, хлорамбуцил, метилнитрозомочевина) до современных (гемзар, флудара, паклитаксел, гливек, ритуксимаб) и даже перспективных (TRAIL, ET-18-OCH3) весьма эффективно активируют программу клеточной смерти. Иными словами, цитотоксины не убивают клетки, а провоцируют их на совершение самоубийства. Несмотря на то что у раковой клетки нарушены функции генетического самоконтроля, лекарства, активирующие программу клеточной гибели, преимущественно поражают все-таки именно клетки опухоли! В этом состоит один из центральных парадоксов химиотерапии опухолей: система распознавания мутаций, поломка которой делает клетку восприимчивой к мутагенезу и ведет к ее опухолевому перерождению, представляет собой лишь часть «молекулярной кухни», реализующей программу клеточной гибели. Факт остается фактом — подавляющее большинство клеточных линий, то есть стационарно поддерживаемых в культуре раковых клеток одного происхождения, используемых для поиска противоопухолевых лекарств, способны к гибели в результате апоптоза.
Однако если действие противоопухолевых лекарств направлено именно на активацию программы клеточной гибели, то следует предположить, что раковая клетка, в которой генетическая программа ее собственной смерти повреждена или вовсе утрачена, должна оказаться устойчивой к действию всех известных препаратов. Доказательство такого предположения неожиданно было получено в нашей лаборатории в Онкологическом центре.
Клетки А4
В самом начале текущего века мы изучали активацию программы клеточной гибели моноклональными антителами к одному из рецепторов смерти, Fas. Этот рецептор появляется на поверхности зрелых лимфоцитов, а также присутствует на некоторых видах злокачественных лимфобластных клеток. Мы использовали моноклональные антитела к этому рецептору, которые имитируют действие природного лиганда FasL, возбуждают рецептор и активируют сигнал клеточного самоубийства. Для экспериментов мы выбрали хорошо известную линию Т-лимфобластных клеток человека Jurkat, выделенных много лет назад из крови больного лейкозом мальчика, на поверхности которых присутствует рецептор Fas. Добавление в питательную среду aнти-Fas-моноклональных антител вызывает быстрое развитие апоптоза этих клеток. Нам нужно было получить культуру, в которой клетки были бы лишены этого рецептора или чтобы рецептор оказался неработающим, то есть клетки, полностью устойчивые к действию анти-Fas-антител. Для этого мы использовали известный прием клеточной селекции, выращивая культуру в присутствии микроскопических концентраций антител. По мере роста культуры концентрацию антител постепенно увеличивали, пока не получили клетки, прекрасно растущие в среде с антителами. Поскольку полученная в результате этого эксперимента культура первоначально росла в чашке с номером А4, мы ее так и назвали — А4, еще не предполагая, что это чисто рабочее название присвоено совершенно уникальной клеточной линии.
По своему внешнему виду и набору поверхностных антигенов клетки А4 сходны с родительскими клетками Jurkat, но не имеют рецептора Fas, поэтому анти-Fas-антитела не стимулируют их гибель. Этот результат не был неожиданностью. Озадачивало другое: полученный клон в полной мере сохранил присущую родительской линии экспрессию рецепторов смерти других типов: АРО-2 для лиганда TRAIL и TNFR-1 для цитокина TNFa, однако применение этих лигандов не вызвало у клеток А4 никаких признаков апоптоза, хотя каждый из них активировал программу гибели родительских клеток Jurkat. Объяснение этому феномену могло быть только одно: устойчивость клеток А4 к апоптозу обусловлена не отсутствием соответствующего рецептора смерти, а нарушениями в каскаде последующих реакций передачи апоптозного сигнала.
Поскольку сигнальные каскады от «внешних» (рецепторы) и «внутренних» (поражение внутриклеточных мишеней) сигналов апоптоза сливаются в общий исполнительный механизм, мы попытались запустить программу клеточного самоубийства, действуя не на внешние рецепторы, а на внутриклеточные триггеры. Для этого клетки обрабатывали цитотоксическими лекарствами разных классов, индукторами окислительного клеточного стресса (перекисью водорода или витамином К3), рентгеновским и ультрафиолетовым излучением. Во всех случаях доля клеток с признаками индуцированного апоптоза в популяции клона А4 оказывалась в 2–10 раз ниже, чем у клеток Jurkat.
Из наших результатов следует, что стимулы самой различной природы, активирующие программу клеточной гибели опухолевых клеток Jurkat, практически не вызывают апоптоз клеток клона А4. Означает ли этот факт, что клетки А4 нельзя убить? Разумеется, нет. Клетки А4 можно умертвить, но такой дозой лекарства, которая несовместима с жизнью пациента. На родительскую линию Jurkat цитостатики действуют в концентрациях на один-два порядка ниже. Другими словами, не способные к апоптозу клетки А4 проявляют фенотип множественной лекарственной устойчивости.
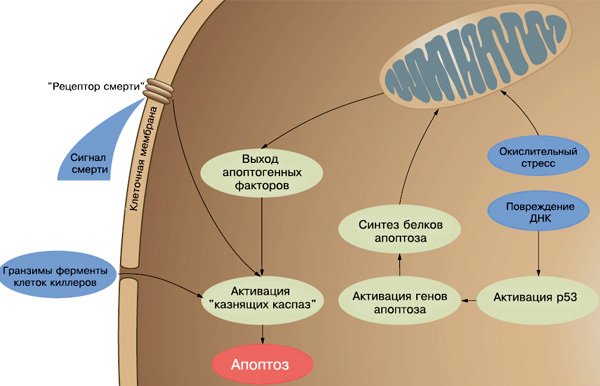
Рис. 3. Программу клеточной гибели могут активировать разные сигналы. Изображение: «Химия и жизнь»
Чтобы выяснить, как клеточная культура реагирует на то или иное лекарство, ее обычно обрабатывают препаратами в концентрации, вызывающей гибель ровно половины клеток (LD50), и наблюдают за судьбой второй половины, пережившей токсическую атаку. В дальнейших исследованиях мы выращивали клетки обеих линий в среде с цитостатиками цисплатином, доксорубицином или этопозидом (каждое из этих лекарств вызывает апоптоз клеток Jurkat). Для клеток А4 концентрации лекарств LD50 были в 30–100 раз выше. Подсчет и морфологический анализ клеток, переживших цитотоксическую атаку, показал, что клетки Jurkat погибают в основном по механизму апоптоза, а клетки А4 — путем некроза, безвременной и неестественной кончины клетки, попавшей в невозможные для жизни условия; их ядро и цитоплазма набухают, а затем разрываются ядерная и клеточная мембраны. Различной оказалась и судьба потомков выживших в этих условиях клеток обеих линий: после пересева в полную питательную среду клетки Jurkatвосстановили исходный облик и скорость роста через три недели, хотя в культуре было еще довольно много умирающих клеток. Потомки клеток А4 даже спустя три недели культивирования продолжали гибнуть в огромном количестве. В их популяции появились как многоядерные клетки, так и клетки с микроядрами — результат неравномерного распределения генетического материала в процессе деления.
Клетки Jurkat, служившие исходным материалом в нашем исследовании, не экспрессируют белок р53, поэтому их геном достаточно изменчив и склонен к накоплению дополнительных мутаций. Вероятно, клетки А4, отобранные из общей популяции в результате Fas-опосредованной селекции, представляют собой клон, появившийся в результате одной или нескольких таких мутаций, природа которых пока не установлена. Собственно, она и не важна и, скорее всего, представляет лишь один из множества возможных вариантов. Важен результат: утратив программу клеточной гибели, клетки А4 получили возможность выжить в присутствии таких высоких концентраций противоопухолевых лекарств, которые больной перенести не может, следовательно — формировать опухолевую ткань, абсолютно устойчивую к лекарственному лечению.
Поскольку клон А4 сформировался спонтанно, можно предположить, что и у онкологических больных клетки, утратившие программу клеточной гибели, могут возникать на разных этапах прогрессии опухоли, независимо от того, какими лекарствами их лечат. И весь имеющийся арсенал специфических противоопухолевых средств оказывается бессильным перед таким клоном.
Эта ситуация — печальное следствие применяемой до настоящего времени методологии отбора новых противоопухолевых лекарств, при которой используют клеточные линии, в большей или меньшей степени сохраняющие способность к программированной гибели. В результате такого скрининга отбирают наиболее эффективные индукторы апоптоза, которые не представляют реальной опасности для опухолевых клеток, утративших к нему способность.
Существует ли выход из тупика? Сегодня на этот вопрос нет ответа. Однако стоит обратить внимание на тот факт, что выжившая после обработки цитотоксинами часть культуры А4 продолжала погибать на протяжении нескольких десятков клеточных делений, происходивших в отсутствие лекарственных препаратов. Этот известный в радиобиологии феномен называется «репродуктивной гибелью», которая наблюдается в том случае, если полученные клеткой генетические повреждения становятся смертельными после одного или нескольких циклов удвоения ДНК. Если же смерть клетки от внешнего воздействия происходит до первого деления, говорят об интерфазной гибели. Почему же «неубиваемые» клетки А4 гибнут без внешних причин? Как это ни парадоксально звучит, причиной их гибели является утрата способности к апоптозу.
«Обычные» раковые клетки сохраняют способность к апоптозу. Поэтому их часть, получившая опасные повреждения, самоликвидируется, но зато оставшиеся клетки продолжают размножаться, как в случае сJurkat, и опухоль живет. А клеткам с утраченной программой гибели ничто не препятствует бесконтрольно накапливать мутации и другие потенциально опасные повреждения, которые могут и не приводить к смерти в интерфазе, но будут препятствовать нормальному процессу клеточного деления и разрушать работу генов, что в конце концов создает ситуацию, несовместимую с дальнейшей жизнью. Поэтому с клетками, на которые не действуют индукторы апоптоза, можно попробовать бороться иным путем: стимулировать образование в них мутаций, чтобы сумма возникших генетических повреждений привела к утрате жизнеспособности их самих и в особенности их потомков. Для этой цели можно попытаться использовать супермутагены или хроническое облучение малыми дозами ионизирующей радиации. Успех будущих исследований во многом зависит от правильного выбора модели для поиска активных соединений.
Мы полагаем, что полученная нами линия клеток А4, а также подобные ей, могут оказаться полезными моделями для поиска принципиально новых противоопухолевых средств, действие которых не ограничится активацией апоптоза. Конечно, существует опасность побочного влияния таких веществ и на клетки нормальных тканей, ведь мутации будут возникать и в их геномах. Но, в отличие от опухолевых, в них продолжает функционировать механизм генетического самоконтроля, не позволяющий тиражировать генетические дефекты в следующих поколениях. Насколько перспективным окажется использование в лечебных целях супермутагенов, покажет будущее.